
以铁依赖性脂质过氧化为标志的铁下垂,可能是癌症治疗的致命弱点。铁下垂抑制蛋白-1 (FSP1)作为铁下垂的第二支柱,通过NAD(P) h依赖性醌的还原有效地防止脂质过氧化。由于其分子机制仍然不清楚,我们研究了许多FSP1突变存在于癌症中或通过非靶向随机诱变确定。该突变分析阐明了FSP1的FAD/NAD(P) h结合位点和质子传递功能,在不同的NADH醌还原酶中具有进化保守性。通过随机诱变筛选,我们揭示了下一代FSP1抑制剂的作用机制。我们的研究确定了第一个FSP1抑制剂iFSP1的结合袋,并引入了第一个独立于物种的FSP1抑制剂,靶向NAD(P) h结合袋。总之,我们的研究为FSP1的分子功能提供了新的见解,并为合理设计针对癌细胞的FSP1抑制剂提供了可能。
自从人们认识到铁死亡是一种以细胞膜氧化破坏为特征的独特的铁依赖性细胞死亡形式以来,这一过程引起了极大的兴趣,可能是因为它与人类疾病,如神经退行性疾病、组织缺血-再灌注损伤和恶性肿瘤有很高的相关性。特别是,某些癌症细胞状态,包括癌症干细胞和治疗耐药和传播的癌细胞,已被报道表现出对铁下垂的固有脆弱性,这为选择性诱导铁下垂作为下一代癌症治疗方法提供了理论依据2,3,4,5,6,7。最近,FSP1被确定为谷胱甘肽过氧化物酶4 (GPX4)的一个重要调控因子8,9,10,11的强大备份系统。通过减少醌,如线粒体外泛醌(CoQ10)或维生素K,以NAD(P)H为代价,NAD(P)H - CoQ10 -维生素K - fsp1轴在磷脂自由基水平上避免脂质过氧化链反应和相关的铁死亡。FSP1在许多癌细胞系中表达,敲除FSP1编码基因凋亡诱导因子线粒体相关2 (Aifm2)对胚胎发育没有影响,也不会在成年小鼠中引起任何明显的表型10,12,13。因此,与GPX4相比,FSP1可能更适合作为肿瘤细胞中的靶标(参考文献)。14,15,16,17),已知它对早期胚胎发生和各种器官(如肾,肝和脑)的组织稳态至关重要18。
尽管FSP1在预防铁下沉中的核心作用已经确立,但该过程背后的详细分子和结构机制仍然难以捉摸,并且对某些癌症类型中发生的FSP1突变的研究仍处于起步阶段。第一个报道的FSP1抑制剂(即iFSP1)8对人类酶具有特异性19,20,这种特异性阻碍了对其精确作用机制(MoA)的深入研究,从而阻碍了对不同FSP1同源物之间的有机体差异和相似性的分析。鉴于这些局限性和不足,我们进行了全面的化学和遗传筛选,以了解FSP1功能的分子和结构基础以及FSP1在细胞铁下沉易感中的作用,并确定可以靶向不同生物体中FSP1酶的FSP1抑制剂。
为了了解FSP1功能的分子和结构基础,我们调查了不同生物中FSP1同源物和烟酰胺腺嘌呤二核苷酸(NADH) -醌还原酶的一致序列和序列基序。其中,II型NADH -醌还原酶(NDH-2s)是一种特征明确的膜蛋白,参与呼吸链21,22,与FSP1一样,是一种能够有效利用NADH还原醌的黄酮类蛋白。NDH-2s含有两个保守的nadh结合基序(GXGXXGXE, WXXG)和一个黄素腺嘌呤二核苷酸(FAD)结合基序(GD)23。有趣的是,FSP1中这些一致的氨基酸序列在物种之间具有很好的保守性(图1a,b和扩展数据图1a,b)。此前,分别位于人类FSP1的GXGXXGXE和GD结合位点的E156A和D285N取代被报道会影响FSP1的酶活性(参考文献)。9,24),尽管G244 (WXXG motif)在FSP1中的功能相关性尚不清楚。此外,在人类癌症体细胞突变数据库中记录了nad结合基序内的一个替换(即p.G244D) (https://cancer.sanger.ac.uk/cosmic)25)(扩展数据图2a,b)。因此,我们首先分析了在体外用天冬氨酸取代G244是否会影响FSP1的酶活性8,10。事实上,G244D取代比E156A取代在更大程度上消除了其氧化还原酶活性(图1c,d)。这种酶活性的丧失与失活的D285N突变体非常相似,D285N突变体缺乏假体,即黄素腺嘌呤二核苷酸(FAD)(图1c,d)。然后,我们利用4-羟基他莫昔芬(Tam)诱导的Gpx4缺失(即Pfa1细胞)26的小鼠胚胎成纤维细胞,以及Gpx4抑制剂(1S,3R)-RSL3 (RSL3)2处理的细胞,研究了这些替换在抑制铁凋亡中的细胞相关性。我们纳入了具有体细胞替换的细胞V148L和S187F,这也在数据库中报道,作为直接比较,以及其他经过充分研究的改变,如G2A和Lyn11膜锚定序列(Lyn11)标记的G2A(扩展数据图2c-f)。已知G2A变体是肉豆铁酰基缺陷突变体,强烈影响FSP1定位,从而取消其铁致死抑制功能8。相比之下,Lyn11-G2A突变体在FSP1-G2A的n端有一个膜靶向序列,因此可以抑制铁凋亡9。尽管其他报道的体细胞替换(V148L和S187F)和Lyn11-G2A突变体在一定程度上使FSP1能够防止铁死亡,但G244D突变体与D285N和G2A突变体一样,完全丧失了其铁死亡抑制功能(图1e,f和扩展数据图2d,e)。为了进一步验证这些共识基序,我们利用了由AlphaFold2(参考文献)预测的FSP1的蛋白质结构。27,28),这与酵母NDH-2酶Ndi1的晶体结构完全一致(参考文献29)(扩展数据图1b),导致FSP1与FAD, NADH和CoQ5的推定结合位点重叠,模拟结构(扩展数据图2g)19。根据这个模型的FSP1结构,G244是nadh结合域的重要组成部分(图1g),类似于其他NDH-2s(扩展数据图1b),进一步支持体细胞取代G244D消除FSP1的铁沉降抑制作用的研究结果。
图1:G244对于FSP1预测的NAD(P) h结合位点至关重要。

a,保守的nadh结合位点(GXGXXGXE和WXXG)。不同物种FSP1的蛋白比对:智人(人类)、小家鼠(小鼠)、褐家鼠(大鼠)、鸡、爪蟾(青蛙)、斑马鱼、酿酒酵母Ndi1 (PDB: 4G73)、热钙杆菌NDH-2 (PDB: 5NA1)、金黄色葡萄球菌NDH-2 (PDB: 4NWZ)生物体旁边的数字表示用于产生蛋白质排列的每个基因的氨基酸范围。b,保守的fad结合位点(GD)。不同物种FSP1的蛋白比对。c,以瑞祖林为底物的FSP1酶活性测定示意图(左)。在指定浓度的野生型(WT) FSP1、FSP1- e156a、FSP1- g244d或FSP1- d285n存在下,reazurin的还原。数据以平均值±s.d表示。96孔板的3个孔,来自3个独立实验中的1个。FL,荧光。d, FSP1酶活性测定示意图,以甲萘醌为底物(左)。在指定浓度的WT FSP1、FSP1- e156a、FSP1- g244d或FSP1- d285n存在下NADH的氧化。数据来自96孔板的单孔,来自3个独立实验中的1个。e, 300 nM RSL3处理24小时后,稳定过表达WT ha标记的人FSP1 (hFSP1-HA)或FSP1突变体的Pfa1细胞的活力。数据以96孔板3个独立实验中的1个的3个孔的平均值±标准差表示。f、在稳定过表达WT hFSP1或其中一种突变体的Pfa1细胞中,用Tam(1μM)处理或不处理72 h,测定其活力。对未使用Tam的各组数据进行归一化处理。数据以96孔板3个独立实验中的1个的3个孔的平均值±标准差表示。P值的计算采用单因素方差分析(ANOVA),随后采用Dunnett多重比较检验(e,f)。g,来自AlphaFold2数据库(https://alphafold.ebi.ac.uk)的预测FSP1结构,具有一致的NADH-和fad结合基序。辅助因子FAD(黄色)、NADH(蓝色)和CoQ5(绿色)从酵母同源物NDH-2 (Ndi1) (PDB: 4G73)的结构中嵌入。预期的氢键是由Pymol生成的。
源数据
To gain further insights into the molecular mechanisms underlying how FSP1 reduces quinones at the expense of NAD(P)H, we established a random mutagenesis screen as an unbiased approach (Fig. 2a). Using error-prone PCR, up to three DNA mutations, on average, were introduced into the FSP1 gene AIFM2, resulting in one to three amino acid substitutions per FSP1 molecule. The resulting pool of genetic material was subsequently cloned into a lentivirus-based expression vector using seamless cloning. This plasmid pool was then transduced into Pfa1 cells with a very low infection ratio (the multiplicity of infection was approximately 0.1), according to genome-wide CRISPR screening approaches performed by our group and others30,31,32, allowing each cell to express one FSP1 mutant. Transduced cells were divided into two groups: one was collected immediately after blasticidin selection, and the other was treated with 500?nM RSL3 for 2 d before collection. After cells were collected, genomic DNA was extracted and AIFM2 was amplified by high-fidelity PCR and subjected to next-generation sequencing (NGS), with the goal of identifying possible mutations. Then, the complete sequence of AIFM2 was analyzed, and the RSL3-treated group was compared with the control group. By calculating the Z score of each DNA sequence and annotating amino acid alterations in FSP1, we retrieved several expected loss-of function mutations that corresponded to residues in the myristoylation motif (MGXXXS)33, the NADH-binding motif (GXGXXGXE, WXXG) and the FAD-binding motif (GD) (Fig. 2b), substantiating the validity of our approach. To further validate the screening results, we cloned the majority of detected mutations and stably expressed them in Pfa1 cells. Indeed, several mutants are sensitive to RSL3- and tamoxifen-induced ferroptosis (Fig. 2c–e and Extended Data Fig. 3a), indicating that they fail to confer the ferroptosis-suppressive function of FSP1. However, without a three-dimensional (3D) structure of FSP1, we cannot formally exclude that a fraction of these mutations not only directly impact the enzymatic activity, but also affect the binding affinity of substrates, expression levels in cells and/or proper protein folding and localization. Nonetheless, to further investigate the role of the aforementioned amino acid residues, we again took advantage of the modeled structure of FSP1 (Extended Data Fig. 2g). According to this model, D41 and C286, located in close proximity to FAD or the predicted GD motif, respectively (Figs. 1b and 2b), are suggested to be important for FAD binding (Fig. 2f). Additionally, our mutagenesis screen identified E160 and K355 as critical amino acid residues for enzyme activity of ferroptosis suppression (Fig. 2b–e). Mechanistically, NDH-2s and other members of the two-dinucleotide binding domains flavoprotein (tDBDF) superfamily, such as dihydrolipoamide dehydrogenases (DLDs), coenzyme A–glutathione reductases (GRs), apoptosis-inducing factor mitochondria associated 1 (AIFM1), ferredoxin reductases (FNRs), nitrite reductases (NIRs), NADH peroxidases and sulfide-quinone oxidoreductases (SQRs), have the glutamic acid residue (Glu) as a catalytic site, like E172 in Staphylococcus aureus (Fig. 2g), as well as a nearby glutamic acid and/or lysine, like K379 in S. aureus34. In line with this mechanism, E156 and K355 of FSP1 are structurally well conserved. Additionally, sequential acidic amino acid residues in close proximity to the glutamic acid have been proposed to be part of a conserved proton-transfer function during quinone reduction23. This proton-transfer function presumably consists of a proton transfer through sequential, conserved carboxylic residues in the α-helix (that is E172, E176, D179 and E183 for S. aureus (Fig. 2g), E169, E173, D176 and E180 for C. thermarum, and E242, E246, D249 and D254 for S. cerevisiae, respectively), and a residue for hydrogen bond formation and subsequent protonation of the quinone upon reduction in the active site (that is, K379 for S. aureus, K376 for C. thermarum, and Y482 for S. cerevisiae, respectively) (Fig. 2g and Extended Data Fig. 3b,c). In accordance with this protonation pathway, E156, E160 and E164 are present in the α-helix, and K355 is in close proximity to the catalytic site in FSP1 (Fig. 2h). Thus, this highly conserved proton-transfer mechanism involving sequential carboxylic acid and lysine residues is most likely crucial for the quinone protonation function of FSP1. Therefore, amino acid mutations identified by random mutagenesis, namely E160K, K355E and K355G, can be considered detrimental because they potentially interrupt the proton-transfer chain. To validate these FAD and proton-transfer functions, we generated corresponding alanine mutants and overexpressed them in Pfa1 cells. Except for the E164A and K43A mutants, all remaining Ala substitutions failed to rescue from ferroptosis induced by either knockout (KO) of Gpx4 or pharmacological inhibition of GPX4, suggesting that these positions are essential for FSP1 function (Fig. 2i and Extended Data Fig. 3d,e).
图2:无偏遗传筛选揭示了FSP1的质子传递函数。

a、突变屏示意图。使用易出错PCR(步骤1)对AIFM2进行突变,然后使用无缝克隆酶“in-Fusion”(步骤2)将PCR片段克隆到质粒(i. Reference)中。使用随机突变质粒的慢病毒池被转导到Pfa1细胞中(ii. Pfa1细胞)。低感染多重性(MOI)的对照(步骤3)。通过RSL3选择过表达突变FSP1的细胞(iii)。收集存活的克隆并提取基因组DNA,然后进行NGS测序(步骤5)。分析测序结果并鉴定突变(步骤6)。gDNA ext.基因组DNA提取。b, Z-score计算后功能失调FSP1替换的代表性总结。肉豆酰化基序(MGXXXS,黄色)、nadh结合基序(GXGXXGXE、WXXG,浅粉色)或FAD结合基序(GD,浅蓝色)的突变残基,以及预测影响质子传递功能(E160和K355,粉色)或FAD结合基序(D41和C286,蓝色)的突变残基。c, 100 nM RSL3处理24 h或不处理24 h稳定过表达hFSP1-HA或突变体FSP1的Pfa1细胞的活力。数据以平均值±s.d表示。一个实验中384孔板(左)或96孔板(右)的3个孔。d,经RSL3处理24小时或未处理24小时后稳定过表达WT hFSP1-HA或突变体FSP1的Pfa1细胞存活率。e,用1μM Tam处理72 h或未处理的Pfa1细胞稳定过表达WT hFSP1或突变体的活力。对未使用Tam治疗的各组数据进行归一化处理(Tam(-))。数据以平均值±s.d表示。来自3个独立实验中的1个的96孔板的3个孔(d,e)。f,与FAD(黄色)和FAD结合残基叠加的hFSP1结构。FAD从金黄色葡萄球菌NDH-2 (PDB: 5NA1)的结构中嵌入,使用Pymol生成预期的氢键。g,金黄色葡萄球菌NDH-2结构(PDB: 5NA1)与辅助因子FAD(黄色)、NADH(蓝色)和CoQ5(绿色)叠加。质子通过α-螺旋上依次羧基残基(即E172、E176、D179和E183)的转移,以及随后K379最终质子化为泛醌的过程用黑色箭头表示。h, FSP1质子通过α-螺旋上依次羧基残基(即E156、E160和E164)转移,并最终由K355质子化为泛醌,用黑色箭头表示。i.用1μM Tam处理72 h或不处理后,在稳定过表达WT hFSP1或突变体的Pfa1细胞中测定活力。对未使用Tam的各组数据进行归一化处理。数据显示为96孔板中3孔的平均值±标准差,来自2个独立实验中的1个。
源数据
我们之前发现了iFSP1,这是第一个FSP1抑制剂,尽管抑制模式和结合位点尚不清楚。首先,我们关注的是人类和小鼠FSP1亚型之间的半最大抑制浓度(IC50)值的内在差异,因为iFSP1对人类蛋白具有特异性19,20。事实上,稳定过表达人类FSP1的Pfa1 Gpx4-KO细胞对iFSP1处理敏感,而表达小鼠FSP1的细胞则对其有抗性(图3a)。为了确定iFSP1可能靶向的氨基酸残基或残基,我们首先一步一步地将所有人类FSP1残基直接改变为它们的小鼠FSP1对应体,并确认它们的表达水平足以通过tam诱导的Pfa1细胞中Gpx4的KO赋予对铁凋亡的潜在抗性(扩展数据图4a,b)。然后用iFSP1处理稳定表达突变的人FSP1酶的细胞24小时。令人惊讶的是,F360L突变体表现出与小鼠FSP1一样对iFSP1治疗的抗性,反之亦然(图3b,c)20。因此,我们假设F360及其周围残基是假定的iFSP1结合袋的一部分。为了进一步确定实际的结合位点,我们进行了随机PCR诱变筛选,以鉴定ifsp1抗性突变体(图3d)。与上述RSL3筛选方法类似(图2a),随机突变的FSP1库在Pfa1细胞中过表达,用RSL3筛选2天。之后,用Tam处理细胞以去除功能失调的FSP1突变体。然后,将TAM/ rsl3选择的细胞用iFSP1处理7 d,建立iFSP1抗性克隆,然后进行NGS分析。经过数据分析和验证,我们确定H48N、T327K和T327R为ifsp1抗性突变体。为了验证,我们在iFSP1处理24小时后检测了表达F360L、H48N、T327K或T327R的Pfa1 Gpx4-KO细胞的生存能力,结果表明它们确实都对iFSP1具有抗性(图3e和扩展数据图4c)。
图3:iFSP1靶向醌结合口袋。

a, Pfa1 Gpx4-KO细胞的活力,稳定过表达hFSP1-HA或iFSP1处理小鼠(mFsp1)-HA 24小时。b, 10μM iFSP1处理24小时后,稳定过表达WT FSP1- ha或FSP1突变体的Pfa1 Gpx4-KO细胞的活力。c、稳定表达hFSP1、hFSP1- f360l、mFsp1或mFsp1- l360f的Pfa1 Gpx4-KO细胞经iFSP1处理24 h后的活力。d,突变屏幕示意图。e,用iFSP1处理24小时后,稳定表达WT FSP1- ha或FSP1突变体的Pfa1 Gpx4-KO细胞的活力。f, iFSP1对FSP1突变体活性影响的代表性剂量-反应曲线,使用重组纯化的hFSP1蛋白(左)。3μM iFSP1或0μM iFSP1对WT FSP1及其突变体活性影响的代表性反应曲线(右)。数据来自96孔板的单孔,来自3个独立实验中的1个。arb.u。,任意单位。g,结合口袋中iFSP1与醌的比较。H48, T327, S364和F360用洋红色突出显示。硅模拟发现了S364和iFSP1之间通过氢键的相互作用,而H48有望稳定其取向。每个交互用橙色虚线表示。h, iFSP1处理24h后稳定过表达WT hFSP1或hFSP1- s364a的Pfa1 Gpx4-KO细胞的细胞活力。数据显示为来自3个独立实验(a - c,e,h)中的1个的96孔板的3孔的平均值±s.d.。
源数据
为了证实iFSP1直接抑制人类FSP1,并且不作用于NAD(P) h - coq10 -维生素K-FSP1轴的上游或下游,我们在大肠杆菌中异种表达了含有H48N, T327K或F360L替代的FSP1,并纯化了重组FSP1变体。我们发现,在经典的FSP1酶检测中,F360L、H48N和T327K突变体对iFSP1具有抗性,证实了iFSP1是人类FSP1的直接抑制剂(图3f和扩展数据图4d)。有趣的是,人类、鸡和爪蟾在360号位置都含有苯丙氨酸(人类FSP1编号),而小鼠、大鼠(褐家鼠)和斑马鱼在对应位置都含有亮氨酸(扩展数据图4e)。此外,人类FSP1-T327S也对iFSP1敏感,它反映了在X. laevis中发现的丝氨酸残基(Extended Data Fig. 4f)。因此,我们选择并检测了F360(人和鸡)和L360(小鼠和大鼠),正如预期的那样,过表达人或鸡FSP1的Pfa1 Gpx4-KO细胞对iFSP1敏感,而表达大鼠或小鼠FSP1的Pfa1 Gpx4-KO细胞对iFSP1有抗性(扩展数据图4,h)。鉴于这些突变体面对预期的膜附着表面和醌结合袋(扩展数据图2g),可以假设iFSP1靶向醌结合位点(图3g)19,20。根据该模型,iFSP1将通过S364和iFSP1之间的氢键以及F360、Y296和iFSP1之间的π -π相互作用与FSP1结合(图3g,h)。
iFSP1不适用于实验性啮齿动物模型的fsp1抑制研究(扩展数据图4)。因此,为了更好地了解FSP1在更广泛的背景下的作用,包括物种特异性差异,除了人类之外,针对FSP1的抑制剂是非常必要的。为此,我们在小鼠表达FSP1的Pfa1细胞中筛选了10,000个药物样小分子文库(图4a),并鉴定出一种化合物,下文称为多功能FSP1抑制剂(viFSP1),这是一种新的人类和小鼠FSP1抑制剂。在稳定表达小鼠Fsp1或人类Fsp1的Pfa1细胞中,用viFSP1处理会导致明显的脂质过氧化和相关的铁致细胞死亡(图4b-d)。viFSP1诱导的细胞死亡被铁沉抑制剂liproxstatin-1 (Lip-1)35挽救,证实了viFSP1确实导致依赖于FSP1存活的细胞的铁沉细胞死亡(Extended Data Fig. 5a)。为了确定viFSP1是否也可以通过靶向FSP1杀死(癌症)细胞,将viFSP1与GPX4的药理抑制剂(RSL3(参考文献36)和JKE-1674(参考文献37))或GPX4的基因缺失共同处理38,39诱导了许多人类和小鼠癌细胞系以及来自不同物种的过表达FSP1的大鼠成纤维细胞(Rat1)和Pfa1细胞的铁死亡(扩展数据图5b-h和扩展数据图6a-j)。为了进行初步的构效关系研究(SAR),除了定制的类似物外,我们还获得了一些市售的viFSP1类似物,表明苯基环周围甲氧基的取代模式影响人/小鼠的选择性。化合物结构和相应的半最大有效浓度(EC50)值如图5c所示。然后,我们在reazurin还原实验中使用重组hFSP1和mFsp1酶研究了这种下一代FSP1抑制剂是否作为FSP1的直接抑制剂8,10(图4e,f)。事实上,在无细胞靶标系统中,hFSP1和mFsp1酶活性对viFSP1同样敏感,并且计算出的IC50值并没有显示人和小鼠酶之间的主要差异(图4f)。因此,这组数据强烈表明,viFSP1是一个直接的物种无关的FSP1抑制剂。
图4:viFSP1作为一种物种无关的FSP1抑制剂的鉴定。
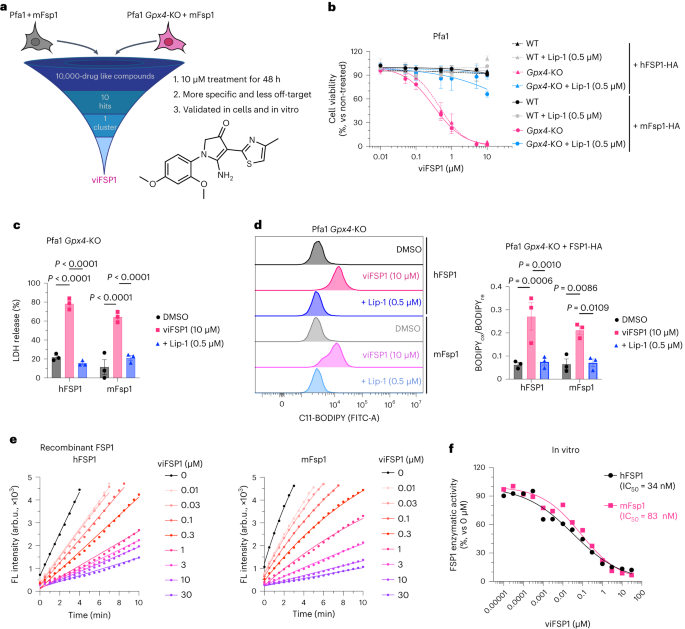
a,化合物筛选鉴定新的mFsp1抑制剂示意图,以及viFSP1的化学结构。Pfa1 Gpx4-KO + mFsp1细胞的存活依赖于mFsp1功能。b, Pfa1 Gpx4-WT和Gpx4-KO细胞在viFSP1处理24小时后稳定过表达人或小鼠FSP1-HA的活力。Lip-1用于抑制铁下垂。c,用DMSO、10μM viFSP1或0.5μM Lip-1处理过表达hFSP1-HA或mFsp1-HA的Pfa1 Gpx4-KO细胞24 h后,测定乳酸脱氢酶(LDH)的释放。d、用DMSO、10μM viFSP1或0.5μM Lip-1稳定过表达hFSP1-HA或mFsp1-HA的Gpx4-KO细胞处理3小时后,通过C11-BODIPY 581/591染色评价脂质过氧化作用。该图代表了三个独立实验(左),并给出了三个独立实验的量化中值(右)。bodipyx,氧化BODIPY的荧光。BODIPYre,还原BODIPY的荧光。数据以平均值±s.e.m表示。三个独立实验(b-d)。P值的计算采用单向方差分析,然后采用Tukey多重比较检验(c,d)。e,不同浓度的viFSP1对hFSP1(左)或mFsp1(右)的抑制效果的代表性分析,使用重组纯化蛋白。f, viFSP1对hFSP1和mFsp1活性影响的代表性剂量-反应曲线。数据来自96孔板的单孔,来自2个独立实验中的1个(e,f)。
源数据
图5:viFSP1靶向FSP1的NAD(P) h结合袋。

a,在hFSP1中确定影响viFSP1活性的相关位点的突变筛选示意图。b,在稳定过表达突变体hFSP1-HA的Pfa1 Gpx4-KO细胞中,用viFSP1处理24小时。数据显示为来自3个独立实验中的1个96孔板的3个孔的平均值±标准差(参见补充视频1 - 4)。c,使用重组纯化的hFSP1蛋白,viFSP1对WT FSP1或其突变体活性影响的代表性剂量-反应曲线(左)。3μM viFSP1或0μM viFSP1对WT FSP1和突变体变体抑制的代表性体外实验(右)。数据来自96孔板的单孔,来自3个独立实验中的1个。d, viFSP1和NADH在各自结合口袋中的比较。A153, G244, M294, T327和F328用洋红色突出显示。硅模拟鉴定了A153、K293和viFSP1主链之间的氢键相互作用。每个交互用橙色虚线表示。
源数据
为了进一步研究viFSP1的竞争作用机制,采用体外实验分析了FSP1酶活性与其底物的关系。利用FSP1也可以以NADH8为代价结合和减少resazurin这一事实,我们计算了在无限浓度的底物下酶催化反应的速度(Vmax)和FSP1抑制剂存在下使用增加NADH或resazurin量的Michaelis常数(Km)(扩展数据图4i和7a)。根据Lineweaver-Burk和Dixon图40,可以推测iFSP1和viFSP1都是非竞争性抑制剂,这意味着这些FSP1抑制剂可以在存在或不存在底物的情况下结合酶;然而,这应该在FSP1的三维结构可用时进行实验研究。
为了更详细地了解viFSP1的抑制机制,我们进行了随机诱变筛选,以鉴定抗viFSP1的突变版本(图5a)。与上述iFSP1筛选类似(图3),通过viFSP1选择稳定过表达随机突变但具有功能的FSP1酶的Pfa1 Gpx4-KO细胞7 d,然后进行NGS分析。经过数据分析和验证,我们确定A153T和F328S为vifsp1敏感突变体,H48N、M294I、M294V、T327K和T327R为vifsp1耐药突变体(图5b和扩展数据图4c和7b)。过表达A153T或F328S的Pfa1 Gpx4-KO细胞对viFSP1处理的敏感性至少是过表达野生型FSP1的细胞的10倍;过表达H48N、M294I、M294V、T327K或T327R变体的细胞对viFSP1处理的抗性大约高出5至10倍(图5b)。除了FSP1的T327K变体外,还使用纯化的重组人FSP1进行了A153T、F328S和M294I变体的体外FSP1酶测定。因此,我们可以观察到在无细胞系统中使用viFSP1抑制FSP1的趋势与在细胞培养中相同,这表明viFSP1是FSP1的直接抑制剂(图5c和扩展数据图7b)。由于这些氨基酸位于NAD(P) h结合位点,并且在物种间高度保守(扩展数据图7c-f),因此可以假设viFSP1靶向NAD(P) h结合口袋(图5d)。
从 

摘要。
主要
结果
讨论
方法
数据可用性
参考文献。
致谢。
作者信息
道德声明
同行评审
扩展数据
补充信息
源数据
这篇文章是由
相关的内容
# # # # #
利用一系列靶向和非靶向化学遗传筛选,我们在这里报道了NAD(P) h结合基元和质子传递函数对FSP1活性的贡献的前所未有的见解(图6),此外还鉴定了一种物种独立的FSP1抑制剂viFSP1。AIFM2基因的一些点突变在癌症患者中以体细胞突变的形式存在,包括编码p.G244D、p.E160D、pE160 stop、p.K355R和p.D285N的点突变(扩展数据图8a)。鉴于这些突变体的功能丧失,携带这些体细胞突变(杂合突变)的癌细胞可能更容易发生铁下垂。鉴于一些癌细胞对GPX4抑制诱导的铁下垂具有抗性,并且FSP1抑制剂使许多癌细胞对亚致死的GPX4抑制诱导的铁下垂敏感,FSP1抑制剂与典型的铁下垂诱导剂(如GPX4和system xc -抑制剂)联合治疗,理想地以肿瘤特异性的方式,可能成为一种新的抗癌治疗方法。Depmap分析(https://depmap.org/portal/, v23Q2)显示,在FSP1扩展数据中,卵巢癌细胞(SKOV3细胞)和子宫内膜/子宫癌细胞(RL952细胞)分别含有G337D和S6L替换。考虑到S6是肉豆肉酰化共识基元的一部分(图2b)33,并且G337D不能防止铁死亡(图2c),这些癌细胞系可能更容易受到铁死亡诱导。
图6:利用FSP1抑制剂观察FSP1及其不同靶点的机制。

利用化合物文库的化学方法和利用靶向和非靶向诱变的遗传方法揭示了两种具有代表性的FSP1抑制剂的作用机制。人类特异性iFSP1靶向醌结合袋,而物种独立的viFSP1靶向NAD(P) h结合袋。此外,一个守恒良好的质子传递函数对FSP1的抗铁猝变活性至关重要。
尽管iFSP1被报道为第一个FSP1抑制剂,并在硅模拟中预测其靶向醌结合位点,但其MoA(包括潜在的结合口袋)仍然不清楚。假设驱动和无偏倚的方法相结合,分别使用位点定向和容易出错的PCR诱变,现在使我们能够确定H48, F360和T327是iFSP结合和抑制人类FSP1所必需的,这在最近的一项独立研究中也有报道20。考虑到这些氨基酸残基中的一些在物种之间可能存在很大差异,这一知识可以用于开发FSP1抑制剂,以实现物种特异性选择性。此外,对FSP1中携带T327M突变的癌症患者的生物信息学分析(扩展数据图6b)25表明,某些个体可能对FSP1抑制剂治疗产生耐药性,因此可能需要个性化的治疗方法。
由于iFSP1对人类FSP1具有特异性,我们在这里引入了一种与物种无关的FSP1抑制剂viFSP1,它将在研究FSP1同源基因的功能时被证明是有用的。值得注意的是,另一组最近报道了基于类似支架的化合物FSEN15(参考文献41),尽管没有详细的SAR分析,也没有对相互作用位点进行全面的表征。无论如何,我们现在表征了viFSP1,它在培养细胞和无细胞系统中更有效,作为一种直接抑制剂,靶向围绕FSP1残基A153, F328, M294和T327的nadh结合袋。
通过使用随机突变筛选和容易出错的PCR来识别FSP1抑制剂的结合袋,我们推断,在寻找相关靶蛋白的结合袋时,本文所述的筛选策略可以应用于其他化合物42。由于人工智能技术的最新进展,当与AlphaFold2和其他对接模拟的预测结构相结合时,药物抑制剂的识别将变得更加准确、直接和可靠。我们证实,在Gpx4基因缺失后,所有对FSP1抑制剂耐药或敏感的突变体确实能使细胞存活,这表明这些突变体一定是功能性的。然而,由于FSP1三维结构的实验数据在这个阶段仍然难以获得,我们不能正式排除氨基酸改变影响FSP1折叠和/或其他未被识别的翻译后修饰的可能性。如上所示,这类信息非常有用;然而,在基于建模方法解释数据时必须谨慎。在不久的将来,需要实验验证FSP1的三维结构。作为展示,我们将这些遗传学和生物信息学方法应用于FSP1,并确定了一系列突变,这些突变最终决定了细胞对铁下垂的敏感性。从突变分析中收集的信息可能在药物遗传学研究中非常有益,以预测未来临床环境中每个患者的铁下垂治疗效果。
利普司他汀-1 (Lip-1: Selleckchem, cat)不。S7699), (1S,3R)-RSL3 (RSL3:开曼,猫。no. 19888), iFSP1 (ChemDiv, cat。不。8009-2626或开曼猫。不。Cay29483), viFSP1 (ChemDiv, cat。不。D715-1847或Vitas M实验室,猫。不。STK626779),甲磺酸去铁胺盐(DFO: Sigma, cat)。不。138-14-7),铁抑素-1 (fe -1: Sigma, cat。不。SML0583), zVAD- fmk (zVAD: Enzo Life Sciences, cat。不。ALX-260-02), necrostatin-1s (Nec-1s: Enzo Life Sciences, cat。不。bv - 2265 -5), MCC950 (Sigma, cat。不。5381200001),奥拉帕尼(Selleckchem, cat;不。S1060)和JKE-1674(开曼,猫。不。Cay30784-1)在本研究中使用。定制的化合物从语调研究实验室获得。
4-羟基他莫昔芬(Tam)诱导的Gpx4 - / -小鼠永生化成纤维细胞(称为Pfa1细胞)先前已有报道26。使用tam诱导的Cre重组酶和CreERT2-LoxP系统可以实现基因组Gpx4的缺失。从ATCC获得HT-1080 (CCL-121)、HEK293T (CRL-3216)、786-O (CRL-1932)、A375 (CRL-1619)、B16F10 (CRL-6475)、LLC (CRL-1642)、MDA-MB-436 (HTB-130)、SW620 (CCL-227)、NCI-H460 (HTB-177)和4T1 (CRL-2539)细胞。SKOV3(91091004)细胞来自Sigma-Aldrich。he151细胞(JCRB1122-A)来自Tebubio。MC38细胞(可从Sigma获得)是P. Agostinis (KU Leuven, Belgium)赠送的。大鼠1细胞(可从赛默飞世尔获得)是汉诺威医学院的礼物。Huh7细胞(可从赛默飞世尔获得)是R. Schneider(亥姆霍兹慕尼黑)的礼物。Pfa1、786-O、A375、SW620、Huh7、HT-1080、Rat1、MC38、LLC和B16F10细胞在含10%胎牛血清(FBS)、2 mM l-谷氨酰胺和1%青霉素-链霉素的dmem -高糖(4.5 g葡萄糖L-1)培养基中培养。将MDA-MB-436、HEC151、SKOV3、H460和4T1细胞培养在含10%牛血清和1%青霉素-链霉素的RPMI GlutaMax培养基中。为了获得稳定过表达FSP1的细胞系,使用抗生素(purromycin 1μg mL-1和blasticidin 10μg mL-1)进行选择。培养gpx4缺陷细胞时,补充1μM Lip-1。所有细胞在37℃、5% CO2条件下培养,证实支原体阴性。
在BL21大肠杆菌中产生重组人和小鼠FSP1蛋白和FSP1突变体,并通过Ni-NTA系统亲和层析纯化。
reazurin实验采用含15-200 nM重组人或小鼠FSP1及其突变体、200 μM NADH和抑制剂(iFSP1和viFSP1)的TBS缓冲液(50 mM Tris-HCl, 150 mM NaCl)制备酶反应。加入100 μM resazurin钠盐(Sigma, cat。不。R7017),荧光强度(FL强度,激发/发射波长(Ex/Em)=540 nm/590 nm)在37°C下使用SoftMax Pro v7 (Molecular devices)的SpectraMax M5或SpectraMaxiD5微孔板读卡器每30或60 s记录一次。不加瑞唑脲(reazurin)的反应归一化并计算FSP1酶活性和IC50值。使用GraphPad Prism v9进行曲线拟合和IC50值计算。
对于nadh消耗测定,酶反应在PBS (Gibco, cat。不。14190094)含有25-200 nM重组人FSP1(或其突变体)和50 μM menadione (Sigma, cat。不。M5625)制备。加入200 μM NADH后,使用SpectraMax M5微孔板读卡器(Molecular Devices)每30 s测量一次在37℃下340 nm处的吸光度。不加NADH或不加酶的反应使结果归一化。曲线拟合使用GraphPad Prism v9完成。
分析酶动力学,反应在PBS (Gibco, cat。不。14190094)含100 nM重组人FSP1和0.04-500 μM NADH与100 μM resazurin钠盐或0.03-100 μM resazurin与200 μM NADH混合;然后,使用SpectraMaxiD5酶标仪(Molecular Devices)在37℃下每60 s记录一次荧光强度(Ex/Em=540/590 nm)。使用GraphPad Prism v9计算Km和Vmax时,采用FL强度的初始斜率。
细胞接种于96孔板(每孔2000 - 10000细胞用于铁下垂诱导剂,每孔500细胞用于TAM)或384孔板(每孔800细胞),培养过夜。第二天,更换培养基,加入如下化合物:RSL3, iFSP1, viFSP1, Lip-1, DFO, Fer-1, zVAD, Nec-1s, MCC950和奥拉帕尼,按图中所示浓度添加。使用AquaBluer (MultiTarget Pharmaceuticals, cat.),在治疗后24-48小时(RSL3、iFSP1和viFSP1)或72小时(TAM)测定细胞活力。No.6015)或0.004% resazurin作为活细胞的指示物。
在正常细胞培养基中孵育4-6小时后,使用带有SoftMax Pro v7 (Molecular devices)的SpectraMax M5酶标仪在Ex/Em=540/590 nm处测量荧光。细胞活力(%)归一化,并在未经处理的条件下计算,没有铁下垂诱导剂或他莫昔芬。
使用MuSyC (https://musyc.lolab.xyz)38,39)评估协同效应。
在LDH释放试验中,每孔2500个细胞接种于96孔板上,培养过夜。第二天,将培养基改为含有抑制剂的新鲜DMEM,再孵育24 h。使用细胞毒性检测试剂盒(LDH) (Roche, cat.)测定坏死细胞死亡。不。11644793001)遵循制造商的协议。简而言之,收集细胞培养上清作为培养基样本,然后用0.1% Triton X-100在PBS中裂解细胞作为裂解物样本。培养基和裂解液样品分别与试剂在96孔板上混合,室温下反应混合物孵育15 - 30min。然后,使用SpectraMax M5酶标仪(Molecular Devices)在492 nm处测量吸光度。以LDH释放量(%)计算细胞死亡率:(培养基样品吸光度(abs)) /(裂解液abs) +(培养基样品abs)) × 100。
实验前一天,每孔10万个细胞接种在12孔板上。第二天,细胞用10μM viFSP1处理3小时,然后用1.5μM C11-BODIPY 581/591 (Invitrogen, cat)孵育。不。D3861)在37°C的5% CO2气氛中放置30分钟。随后,用PBS洗涤细胞一次,在37°C下胰蛋白酶化。然后,将细胞重悬于500μL PBS中,通过40 μm细胞滤网,然后使用流式细胞仪(CytoFLEX,软件(CytExpert v2.4), Beckman Coulter),在488 nm激光激发下进行分析。采用525/40 nm带通滤波器的异硫氰酸荧光素(FITC)检测器(用于氧化形式的BODIPY)或采用585/42 nm带通滤波器的藻红蛋白(PE)检测器(用于还原形式的BODIPY)收集数据。每个样本至少分析了10,000个事件。使用FlowJo软件对数据进行分析。使用每个通道的中位数计算C11-BODIPY 581/591(脂质过氧化)的荧光比率(FITC/PE比率(氧化/还原))30。
Pfa1细胞(5000 - 10000个)接种于35mm, low (ibidi, cat)μ-Dish上。不。80136),孵育过夜。第二天,将细胞培养基改为新鲜培养基。使用3D Cell Explorer (Nanolive)软件Evev1.8.2进行活细胞成像。在成像过程中,使用温度控制的孵育室将细胞保持在37°C和5% CO2气氛中。记录一张图像后,在培养皿中加入100倍浓度的DMEM viFSP1(终浓度为10μM viFSP1),然后继续记录。为了抑制铁下垂,细胞在记录前预处理0.5μM Lip-1 15分钟。每5分钟拍照一次,持续6小时以上。
细胞在LCW裂解缓冲液(0.5% Triton X-100, 0.5%脱氧胆酸钠盐,150 mM NaCl, 20 mM Tris-HCl, 10 mM EDTA, 30 mM na -焦磷酸四碱十水)中裂解,并添加蛋白酶和磷酸酶抑制剂混合物(cOmplete和phoSTOP, Roche, cat)。不。04693116001和cat。不。4906837001),并在20000 g, 4°C下离心30分钟至1小时。细胞裂解液用6×SDS上样缓冲液(375 mM Tris-HCl, 9% SDS, 50%甘油,9% β-巯基乙醇,0.03%溴酚蓝,pH 6.8)溶解。在95°C煮沸3分钟后,样品在12% SDS-PAGE凝胶(Bio-Rad, cat)上溶解。不。4568043或猫。不。4568046),随后电印迹到聚偏二氟乙烯(PVDF)膜上(Bio-Rad, cat)。不。1704156或cat。不。1704274),遵循制造商的协议。膜在阻断缓冲液中孵育,5%牛奶(Roth, cat;不。T145.2)在TBS-T (20 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20)中检测,然后用一抗检测。一抗在抗体稀释缓冲液中稀释(5% BSA, 0.1% NaN3 (Sigma, cat。no.S2002)在TBS-T)和GPX4 (1:1000, Abcam, cat。不。ab125066),含缬草苷蛋白(VCP, 1:10 000, Abcam, cat)。不。ab109240), HA标签(1:10 000,大鼠IgG1,克隆3F10,内部开发),人类FSP1 (1:10 000, Santa Cruz,猫。不。sc-377120, AMID)或人FSP1(1:5,大鼠IgG2a,克隆AIFM2 6D8,内部开发),人和小鼠FSP1(1:5,大鼠IgG2a,克隆AIFM2 1A1-1,内部开发),人和小鼠FSP1(1:5,大鼠IgG2b,克隆AIFM2 14D7,内部开发),或在5%牛奶中稀释TBS-T抗山根过氧化酶偶联β-肌动蛋白(1:5万,Sigma,猫)。不。一夜之间A3854)。膜清洗一次后,用二抗(1:10 000 - 1:5 000,Cell Signaling, cat)进行探针。不。7074S为兔子;猫。不。7076S(小鼠)和1:10 00抗大鼠IgG1b和2a/b(自行开发)在TBS-T中稀释5%脱脂牛奶。抗体-抗原复合物的检测采用ChemiDoc成像系统与Image Lab v6.0 (Bio-Rad)。使用ImageJ/Fiji软件(ver.1.53)调整到适当的亮度和角度后显示代表性图像。
本研究的质粒均采用标准分子生物学技术构建,并通过测序进行验证:将人AIFM2 cDNA (NM_001198696.2, C>T:1008)和密码子优化的小家鼠(小鼠)FSP1 (NP_001034283.1)、褐家鼠(大鼠)FSP1 (NP_001132955.1)和鸡(鸡)FSP1 (XP_421597.1)克隆或合成为p442- irs -blast载体8,17。为了产生突变体或亚克隆,DNA首先由KOD One (Sigma, cat)扩增。不。KMM-201NV), PCR产物由Wizard SV Gel&PCR cleanup System (Promega, cat)纯化。no.A9285)。用In-Fusion克隆酶(Takara Bio, cat.)将PCR产物与酶切载体进行连接反应。不。639649或638948)。随后,将反应混合物转化为NEB稳定的能态细胞(NEB, cat。no.C3040H)。质粒采用QIAprep Spin Miniprep Kit (QIAGEN, cat)分离。No.27106),然后进行测序。
HEK293T细胞用于产生慢病毒颗粒。小鼠白血病病毒(MLV)的亲生态包膜蛋白用于小鼠源性细胞。第三代慢病毒包装系统由转移质粒、包膜质粒(pEcoEnv-IRES-puro)或pHCMV-EcoEnv (Addgene, cat)组成。no.15802)(生态颗粒))和包装质粒(pMDLg_pRRE和pRSV_Rev,或psPAX2 (Addgene, cat)。no.12260))使用转染试剂(PEI MAX (Polysciences, cat。不。24765)或X-tremeGENE HP制剂(Roche, cat.)。不。06366236001))。转染后2-3 d收集含有病毒颗粒的细胞培养上清,并通过0.45μm PVDF过滤器(Millipore, cat)过滤。不。SLHV033RS),然后在?80°C保存。
细胞用慢病毒外加10μg mL-1硫酸鱼精蛋白在12孔或6孔板上接种过夜。第二天,将细胞培养基替换为含有适当抗生素的新鲜培养基,如purromycin (Gibco, cat。不。A11138-03;1μg mL-1)和杀胚素(Invitrogen, cat.)不。A1113903;10μg mL-1),选择转导的细胞,直到未转导的细胞完全死亡。
利用GeneMorph II随机突变试剂盒(Agilent cat.)从病毒表达载体p442- hfsp1 - ires - blast8中扩增出带有p442适配序列的突变AIFM2 cDNA (NM_001198696.2, C>T:1008)。不。200550)。PCR在以下条件下获得最佳诱变率(每个基因3 - 5个DNA突变):(1)95°C 120 s,(2) 95°C 30 s,(3) 60°C 30 s,(4) 72°C 75 s,(5) 72°C 10 min;步骤2-4的循环重复25次,其他步骤均执行一次。使用琼脂糖电泳分离PCR产物并使用Wizard SV凝胶和PCR清理系统进行清理后,使用In-Fusion酶(Takara, cat)将FSP1片段连接到酶切的p442-IRES-blast载体上。不。639649)在以下条件下:400 ng DNA插入物,200 ng载体,8μL酶,40μL反应体积,50°C, 15 min。然后将插入-载体混合物转化为NEB稳定细胞,37℃孵育1 h。诱导耐药基因后,将细胞置于8个培养皿中(Thermo Fisher, cat;不。240835),用氨苄青霉素选择培养基在30℃下培养过夜。第二天收集菌落,使用QIAGEN质粒Maxi Kit (QIAGEN, cat)分离DNA。不。12163)。
如上所述,使用转染试剂(X-tremeGENE HP agent)将由FSP1突变文库质粒、包膜质粒(pEcoEnv-IRES-puro)和包装质粒(pMDLg_pRRE和pRSV_Rev)组成的第三代慢病毒包装系统共脂转染HEK293T细胞。转染后2 d收集含有病毒颗粒的细胞培养上清,用0.45μm PVDF过滤器过滤,保存于- 80°C。
用含有10 μg mL-1鱼精蛋白和含有FSP1突变体的慢病毒的培养基将Pfa1细胞接种到10个T-175烧瓶中(每个烧瓶1.0 × 106个细胞,总共2.0 × 107个细胞),转染效率极低(MOI=约0.1,按前面描述计算43)。第二天,将培养基替换为含有杀胚素(12.5μg mL-1)和嘌呤霉素(1μg mL-1)的新鲜培养基。用杀胚素筛选3 d后,取1000万个细胞作为对照组;同时,用500 nM RSL3将2000万个细胞接种于5个烧瓶中(每个烧瓶2.0 × 106个细胞,共2.0 × 107个细胞)。RSL3诱导铁下垂2 d后,收集1000万个细胞作为RSL3处理组。然后,每组1000万个细胞分别用单个FSP1抑制剂(前5天5μM,之后10μM)和500 nM RSL3和1μM 4-OH Tam处理,使Pfa1细胞对FSP1抑制剂完全耐药,同时仍然表达功能性的FSP1。然后,扩增过表达耐药FSP1突变体的Pfa1 Gpx4-KO存活细胞,每种条件下收集1000万个细胞,保存在?80°C。
收集所有条件下的细胞后,用蛋白酶K (100 μg mL-1)在裂解缓冲液(50 mM Tris, 50 mM EDTA, 1% SDS, pH 8)中裂解细胞,55°C过夜。第二天,加入RNase A(50μg mL-1), 37℃孵育30 min,消化RNA。然后,苯酚:氯仿:异戊醇的当量体积(25:24:1)(罗斯,猫。不。添加A156.2)。将溶液在16000 g下短暂旋转并离心10分钟,将DNA与RNA和蛋白质分离。在新管中仔细收集含DNA溶液的上相,用2倍体积的75 mM NaCl在16000 g乙醇中沉淀DNA,离心10分钟。然后用70%乙醇洗涤颗粒,然后离心。将剩余乙醇干燥后,将球团溶解于200 μL TE缓冲液中,在65℃下孵育1 h。最后,AIFM2区域的DNA(约1500个碱基对)被KOD One (Sigma, Cat.)扩增。不。KMM-201NV),并按上述方法纯化。
NGS文库制备使用ThruPLEX DNA-Seq HV PLUS试剂盒(Takara, cat)。不。R400782),并进行了少量优化。文库制备完成后,使用NucleoMag NGS cleanup和Size Select (Th)对DNA进行纯化。盖尔,猫。不。11833159),以供日后的NGS查阅。NGS由位于慕尼黑亥姆霍兹的核心设施进行。
使用Illumina NovaSeq 6000仪器,使用ThruPLEX DNA-Seq HV PLUS试剂盒,在不同条件下进行配对端测序(见补充表1)。FastQC (v0.11.7) (http://www.bioinformatics.babraham.ac.uk/projects/fastqc)程序应用于结果FASTQ文件,以识别过度代表的序列(Illumina适配器),并将其排除在进一步分析之外。我们使用Trimmomatic V.039工具44和以下选项(ILLUMINACLIP:TruSeq2-PE_extended)。fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36)来修剪成对端数据(见补充表1)。使用Burrows-Wheeler比对工具(BWA),版本0.7.17-r1188(参考文献45),将修剪后的成对端reads(原始长度为151 bp)与参考AIFM2 cDNA序列1,210 bp进行比对。首先,使用命令' bwa index '为引用序列生成索引。然后,通过应用子命令' mem ', BWA以SAM(序列对齐/映射)格式输出最终对齐。将对齐的reads转换为BAM (Binary Alignment Map)格式,并使用SAMtools version 1.2(参考文献46)程序分别使用“SAMtools view”和“SAMtools sort”命令按最左边的染色体坐标排序。利用整合基因组学查看器(IGV)的IGVtools,版本2.11.9(参考文献47),从排序好的BAM文件中提取FSP1参考序列的每个碱基的覆盖信息。命令' igvtools count '和选项(-w 1和base)用于为每个排序的BAM文件生成WIG (wiggle)格式的输出文件。FSP1参考序列每个位置的突变频率(Xi)由参考序列对应位置的突变核苷酸总和除以该位置所有核苷酸(A,C,G,T,N)的总和计算。存储在摆动文件中的每个碱基计数信息作为自定义编写的R脚本的输入,用于计算突变频率。R脚本的以制表符分隔的输出文件包含每个核苷酸的数量、缺失和插入的数量,以及所有核苷酸的总和和FSP1序列的每个位置(行)的对齐(列)的突变频率。
其中Xi为AIFM2的位置,Nmut为Xi处突变核苷酸的和,Nall为Xi处所有核苷酸的和。用平均值(μ)和标准差(σ)计算Z分数如下:
其中iFSP1表示iFSP1或viFSP1, ctrl表示控件。然后,利用密码子表(补充表1),根据可能的DNA交替来研究氨基酸替换。
将稳定过表达小鼠FSP1的Pfa1和Pfa1 Gpx4-KO细胞分别接种在384孔板上(每孔500个细胞),并用小分子抑制剂化合物文库进行筛选,如先前报道8。用AquaBluer处理后48 h评估不同细胞系的活力。如上所述,在稳定过表达小鼠FSP1的Pfa1 Gpx4-KO细胞中显示选择性致死的化合物随后在细胞活力和体外FSP1酶测定中得到验证。
建立了人GPX4、小鼠GPX4和小鼠Fsp1的单导rna (sgRNA)载体,并生成KO细胞,如先前报道10,17。使用X-tremeGENE HP将表达lentiCRISPRv2-blast或lentiCRISPRv2-puro的sgrna短暂共转染SW620 GPX4-KO细胞。转染1天后,用purromycin(1μg mL-1)和blasticidin(10μg mL-1)选择细胞,直到未转染的细胞死亡。连续稀释分离单细胞克隆,免疫印迹法确认KO克隆。
用X-tremeGENE HP试剂转染4T1 Gpx4-KO细胞和B16F10 Gpx4-KO Fsp1-KO (DKO)细胞141-IRES-puro、141-hFSP1-IRES-puro、141-mFsp1-IRES-puro、141-mGpx4- ires -puro或141-mGpx4 U46C-IRES-puro载体。转染后1天,选择细胞并在puromycin(1μg mL-1)存在和Lip-1不存在的情况下培养,以选择稳定表达FSP1或gpx4的细胞。
从AlphaFold2数据库(https://alphafold.ebi.ac.uk)28)中获得了预测的人类FSP1结构。为了得到FSP1与其辅助因子黄素腺嘌呤二核苷酸(FAD)、NADH和泛醌的重叠结构,使用Pymol v2.5.2 (Schr?dinger)将FSP1同源物NDH-2 (PDB: 4G73 ref. 29或5NA1 ref. 48)的结构与FSP1对齐,提取FAD、NADH和泛醌的位置并嵌入到FSP1结构19或另一个NDH-2 (PDB: 4NWZ ref. 49)中。利用建模软件SeeSAR v12.1 (BioSoveIT)19对iFSP1和viFSP1进行计算机建模。目测后选择最可行的姿态,对接分子导出为PDB文件。所有对接或叠加结构的描绘都是用Pymol绘制的。
通过UniProt (https://www.uniprot.org)、NCBI (https://www.ncbi.nlm.nih.gov/gene/)和PDB (https://www.rcsb.org)获取人类FSP1序列及其同源物,然后使用JalView50 (v2.11.2.6)进行比对和可视化。
使用DNAzol (Fisher Scientific, cat.)提取SKOV3的基因组DNA。No.15413379),根据制造商的说明。测序由Eurofins genomics进行。
所有数据均为平均值±s.e.m。或平均值±s.d。,每个图例中的数字(n)代表生物复制或技术复制。所有实验(除突变分析外)至少独立进行两次。使用GraphPad Prism 9 (GraphPad Prism)进行单因素或双因素方差分析,并进行Dunnett或Tukey多重比较检验(详见图例)。统计分析的结果在每个图中表示。P < 0.05为差异有统计学意义。未采用统计学方法预先确定样本量。没有数据被排除在分析之外。实验不是随机的。研究人员在实验或结果评估期间没有对分配进行盲法。
有关研究设计的更多信息可在本文链接的自然组合报告摘要中获得。
ccDownload: /内容/ pdf / 10.1038 / s41594 - 023 - 01136 - y.pdf








